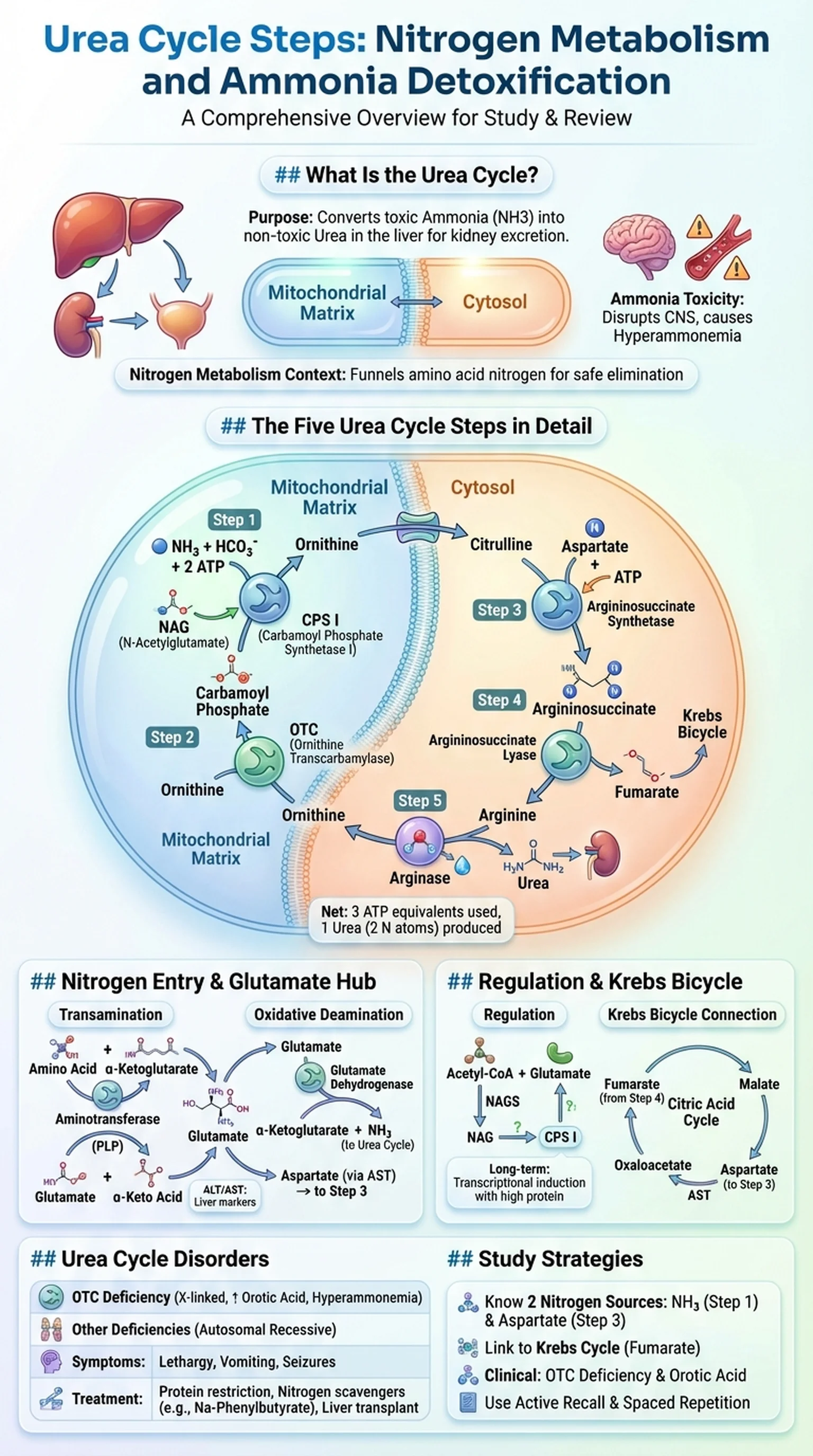
What Is the Urea Cycle?
The urea cycle is a metabolic pathway that converts toxic ammonia into urea, a water-soluble and relatively non-toxic molecule that is excreted by the kidneys in urine. Also known as the ornithine cycle, it was the first cyclic metabolic pathway to be discovered, described by Hans Krebs and Kurt Henseleit in 1932. The urea cycle takes place primarily in the liver, with the first two reactions occurring in the mitochondrial matrix and the remaining three reactions occurring in the cytosol of hepatocytes.
The urea cycle is essential for ammonia detoxification in humans and other ureotelic organisms. Ammonia is produced continuously through the normal catabolism of amino acids, which occurs when proteins are broken down for energy or when excess dietary amino acids are deaminated. Free ammonia is extremely toxic to the central nervous system because it disrupts the glutamate-glutamine balance in astrocytes, depletes alpha-ketoglutarate from the citric acid cycle, and impairs oxidative energy metabolism in neurons. Even modest elevations in blood ammonia can cause confusion, lethargy, and cerebral edema, while severe hyperammonemia can be fatal.
Nitrogen metabolism is the broader biochemical context in which the urea cycle operates. When amino acids are catabolized, their amino groups must be safely collected, transported, and eliminated. The processes of transamination and oxidative deamination funnel nitrogen from various amino acids into glutamate and then into ammonia or aspartate, the two nitrogen donors of the urea cycle. Each molecule of urea contains two nitrogen atoms: one derived from free ammonia and one from aspartate. Understanding the urea cycle is therefore foundational for students studying nitrogen metabolism, amino acid catabolism, and the clinical consequences of metabolic liver disease.
Key Terms
A cyclic metabolic pathway in the liver that converts toxic ammonia and aspartate-derived nitrogen into urea for excretion by the kidneys.
The biological process of converting toxic free ammonia (NH3/NH4+) into the non-toxic, water-soluble waste product urea through the urea cycle.
The collective biochemical processes involved in the assimilation, interconversion, and elimination of nitrogen-containing compounds, including amino acid catabolism and the urea cycle.
An abnormally elevated concentration of ammonia in the blood, often caused by urea cycle defects or liver failure, leading to neurological dysfunction.